

Katarína Furmanová, Jan Byska, Eduard Gröller , Ivan Viola, Jan J. Paleček, Barbora Kozlikova
, Ivan Viola, Jan J. Paleček, Barbora Kozlikova
COZOID: contact zone identifier for visual analysis of protein-protein interactions
BMC Bioinformatics, (19:125 ):1-25, April 2018. [image] [ Paper]
Paper]
Information
- Publication Type: Journal Paper (without talk)
- Workgroup(s)/Project(s): not specified
- Date: April 2018
- DOI: 10.1186/s12859-018-2113-6
- Journal: BMC Bioinformatics
- Number: 19:125
- Open Access: yes
- Pages: 1 – 25
Abstract
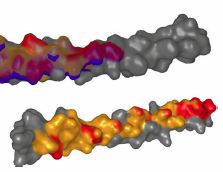
Background: Studying the patterns of protein-protein interactions (PPIs) is fundamental for understanding thestructure and function of protein complexes. The exploration of the vast space of possible mutual configurations ofinteracting proteins and their contact zones is very time consuming and requires the proteomic expert knowledge.Results:In this paper, we propose a novel tool containing a set of visual abstraction techniques for the guidedexploration of PPI configuration space. It helps proteomic experts to select the most relevant configurations andexplore their contact zones at different levels of detail. The system integrates a set of methods that follow and supportthe workflow of proteomics experts. The first visual abstraction method, the Matrix view, is based on customizedinteractive heat maps and provides the users with an overview of all possible residue-residue contacts in all PPIconfigurations and their interactive filtering. In this step, the user can traverse all input PPI configurations and obtain anoverview of their interacting amino acids. Then, the models containing a particular pair of interacting amino acids canbe selectively picked and traversed. Detailed information on the individual amino acids in the contact zones and theirproperties is presented in the Contact-Zone list-view. The list-view provides a comparative tool to rank the best modelsbased on the similarity of their contacts to the template-structure contacts. All these techniques are interactivelylinked with other proposed methods, the Exploded view and the Open-Book view, which represent individualconfigurations in three-dimensional space. These representations solve the high overlap problem associated withmany configurations. Using these views, the structural alignment of the best models can also be visually confirmed.Additional Files and Images

Weblinks
BibTeX
@article{Furmanova_2018,
title = "COZOID: contact zone identifier for visual analysis of
protein-protein interactions",
author = "Katar\'{i}na Furmanov\'{a} and Jan Byska and Eduard
Gr\"{o}ller and Ivan Viola and Jan J. Pale\v{c}ek and
Barbora Kozlikova",
year = "2018",
abstract = "Background: Studying the patterns of protein-protein
interactions (PPIs) is fundamental for understanding
thestructure and function of protein complexes. The
exploration of the vast space of possible mutual
configurations ofinteracting proteins and their contact
zones is very time consuming and requires the proteomic
expert knowledge.Results:In this paper, we propose a novel
tool containing a set of visual abstraction techniques for
the guidedexploration of PPI configuration space. It helps
proteomic experts to select the most relevant configurations
andexplore their contact zones at different levels of
detail. The system integrates a set of methods that follow
and supportthe workflow of proteomics experts. The first
visual abstraction method, the Matrix view, is based on
customizedinteractive heat maps and provides the users with
an overview of all possible residue-residue contacts in all
PPIconfigurations and their interactive filtering. In this
step, the user can traverse all input PPI configurations and
obtain anoverview of their interacting amino acids. Then,
the models containing a particular pair of interacting amino
acids canbe selectively picked and traversed. Detailed
information on the individual amino acids in the contact
zones and theirproperties is presented in the Contact-Zone
list-view. The list-view provides a comparative tool to rank
the best modelsbased on the similarity of their contacts to
the template-structure contacts. All these techniques are
interactivelylinked with other proposed methods, the
Exploded view and the Open-Book view, which represent
individualconfigurations in three-dimensional space. These
representations solve the high overlap problem associated
withmany configurations. Using these views, the structural
alignment of the best models can also be visually confirmed.",
month = apr,
doi = " 10.1186/s12859-018-2113-6",
journal = "BMC Bioinformatics",
number = "19:125 ",
pages = "1--25",
URL = "https://www.cg.tuwien.ac.at/research/publications/2018/Furmanova_2018/",
}